Method development and validation of regulatory qPCR and dPCR assays for biodistribution, viral shedding, and pharmacokinetics in gene and RNA therapies.
qPCR and dPCR in bioanalysis
Quantitative PCR (qPCR) and digital PCR (dPCR) are highly sensitive techniques for detecting nucleic acids, making them ideal for bioanalytical applications such as biodistribution, viral shedding, and pharmacokinetics in gene therapies, RNA therapeutics, and RNA vaccines.
Detecting and quantifying the administered or transgene-expressed nucleic acid can be like finding a needle in a haystack, requiring deep expertise, strategic assay design, and well-placed controls at every step of method development to ensure that the results truly reflect biological reality.
From our experience, factors such as target length, secondary structure, GC content, and chemical modifications significantly influence extraction yield, reverse transcription (RT) efficiency, and PCR performance. Our scientists are experts at interpreting assay behavior and can immediately identify when amplification curves deviate from expectations, enabling fast, efficient, and reliable method development and validation.
While regulatory guidelines for validating qPCR and dPCR assays in bioanalysis are still evolving, we follow the most up-to-date industry best practices, including the 2024 AAPS recommendations, which we co-authored alongside leading experts in the field.
The complexities of biodistribution, shedding, and pharmacokinetic (PK) analysis of RNA and DNA analytes
Understanding where a viral vector, RNA therapeutics, engineered cell, or RNA vaccine ends up in the body is essential for assessing biodistribution, persistence, and clearance, key factors that inform dosing strategies and determine overall therapy success.
For small molecules, pharmacokinetics (PK) focuses on how the drug is absorbed, distributed, metabolized, and excreted, including parameters like peak concentration and duration of action. In contrast, advanced therapies involve a more complex PK profile, as the therapy is not delivered as a ready-to-act drug but initiates a series of biological events post-administration, including cell binding, internalization, transgene expression, and protein translation. In AAV therapies, PK and biodistribution analytes often include the vector genome DNA or transgene mRNA in blood for PK assessments and target and non-target tissues for biodistribution studies.
Viral shedding assessments are required for virus-based gene therapies and oncolytic viral products to evaluate the potential release of the therapeutic vector into the environment through saliva, feces, urine, or other excreta.
Detecting the therapeutic agent, whether mRNA, vector DNA, transgene DNA, siRNA, or other nucleic acid, demands highly sensitive and robust analytical assays. The analytes are often present in very low copy numbers, and in some cases, the objective is simply to determine the presence or absence of the therapeutic drug. However, the analysis is challenging. Tissues and biofluids are complex matrices rich in endogenous DNA and often contain PCR inhibitors. Shedding samples, such as feces, saliva, and urine, are particularly complex, containing organic matter, host proteins, and environmental microorganisms, all of which can interfere with PCR reaction, hence the detection and complicate the interpretation of shedding data.
The sensitivity, i.e., the limit of detection (LOD) and limit of quantification (LOQ) for qPCR, is typically around 10 copies. In comparison, digital PCR (dPCR) can detect as few as 1–2 copies per reaction.
Low variability and a robust assay are essential to validate a method at this sensitivity level. To meet these assay requirements, careful optimization of each step, including PCR efficiency, primer and probe specificity, and assay sensitivity, is needed. A well-optimized and consistent assay is crucial to meet the stringent criteria for full method validation in regulated studies.
Method development
Regulatory guidelines for analytical validation in RNA and DNA bioanalysis
The FDA published the ICH M10 Bioanalytical Method Validation Guidance for Industry in 2022, providing a framework for validating bioanalytical methods used to support regulatory submissions. The guidance emphasizes that validation is essential to confirm that a bioanalytical method performs reliably and produces reproducible results. A full validation should assess selectivity and specificity to ensure the method accurately measures the analyte of interest, evaluate matrix effects to understand how biological matrices influence quantification, and include a calibration curve to establish the relationship between analyte concentration and signal response. The lower and upper limits of quantification (LLOQ and ULOQ) must be defined to determine the assay’s quantification range. Additional requirements include evaluating accuracy, precision, carryover, and analyte stability under various conditions.
The ICH M10 guidelines were developed primarily for conventional bioanalytical techniques such as LC-MS/MS and ligand binding assays, which directly quantify the concentration of analytes in biological samples. In contrast, quantitative PCR (qPCR) determines analyte levels indirectly by measuring the cycle threshold (Ct) value and comparing it to a standard curve generated from known concentrations. Within its validated dynamic range, qPCR exhibits a log-linear relationship between Ct and the logarithm of analyte concentration. However, its accuracy and precision depend highly on consistent amplification efficiency and the absence of PCR inhibitors.
Unlike small molecule analytes, nucleic acids are inherently unstable and susceptible to degradation, requiring stringent quality control measures, including internal spike-ins, degradation assessments, and reverse transcription (RT) controls (for RNA targets). Furthermore, defining the lower limit of quantification (LLOQ) in qPCR requires empirical validation, as it is influenced by variability at low template concentrations and assay efficiency.
Specificity in qPCR assays is determined by the primers and probes’ design and the target sequence’s uniqueness, which presents different selectivity challenges compared to LC-MS/MS. These methodological differences highlight the need to adapt bioanalytical validation principles when applying M10-compliant workflows to nucleic acid-based assays.
The ICH S12 guideline on nonclinical biodistribution provides standardized sample timing, group size, and tissue collection recommendations. The guideline highlights the importance of assessing both the presence and quantity of vector DNA, gene expression products, and/or cellular markers in target and non-target tissues. These tissues include but are not limited to the injection site, gonads, adrenal glands, brain and spinal cord, liver, kidneys, lungs, heart, spleen, blood, and other relevant organs depending on the administration route and therapeutic target. qPCR is highlighted in the guideline as a sensitive technique capable of detecting low copy numbers of nucleic acids, making it an ideal tool for measuring the persistence of gene therapy products in biodistribution studies.
The S12 guideline also emphasizes the importance of screening for pre-existing immunity to the viral vector prior to administration, as pre-existing antibodies or immune responses can alter the biodistribution profile by accelerating vector clearance and degrading the therapeutic agent.
High data quality, integrity, and reliability are essential for regulatory acceptance. While studies not conducted under Good Laboratory Practice (GLP) compliance may be acceptable, GLP becomes critical when biodistribution evaluations are integrated into GLP-compliant toxicology studies. In such cases, all in-life evaluations and sample collections must adhere to GLP standards.
The extent of analytical validation required should be based on a risk-based approach, meaning that the level of validation should reflect the potential impact and regulatory importance of the data generated. For example, assays supporting pivotal safety decisions in regulatory filings may require more rigorous validation than those used in exploratory research.
In 2015, the FDA issued guidance on shedding studies for virus- and bacteria-based gene therapies and oncolytic products. Shedding assays are required to determine whether a therapeutic product releases viral particles or genetic material into bodily fluids, a key consideration for environmental risk and potential transmission. Recommended matrices for shedding assessments include urine, saliva, feces, blood, and other relevant body fluids. At least one assay should be quantitative, allowing results to be expressed in terms of genome copies or infectious units. Quantitative PCR (qPCR) is recommended due to its sensitivity, ease of standardization, high throughput, and rapid turnaround time. The FDA requires that such assays be specific, sensitive, reproducible, and accurate, with clearly defined LOD and LOQ and well-characterized specificity to avoid false positives or false negatives. However, the agency does not expect shedding assays to be fully validated. Instead, they should be qualified to meet minimum performance requirements and be fit for their intended purpose.
Overall, the regulatory landscape for nucleic acid-based bioanalytical assays such as qPCR and dPCR is still evolving. While ICH M10, S12, and the shedding guidelines set a foundation, they do not fully capture the unique characteristics of nucleic acid assays. We therefore refer to current best practices, including recommendations published by AAPS in 2024, which were developed by 37 experts from 24 organizations. These recommendations help fill current regulatory gaps. The following section will discuss the specific validation criteria for qPCR and dPCR-based assays.
Assay development and validation strategies
Absolute quantification using qPCR or dPCR measures the copy number of a target in a sample. In qPCR, this is done by comparing the Ct value of the unknown sample to a standard curve generated from samples with known copy numbers. In dPCR, absolute quantification is achieved without a standard curve by partitioning the sample and directly counting the number of positive reactions. Relative quantification with qPCR measures the fold change in gene expression between samples, often normalized to a reference gene, and is primarily used in gene expression analysis. Absolute quantification is widely applied in bioanalysis for advanced therapies, including biodistribution, pharmacokinetics (PK), and viral shedding assays.
PCR efficiency is a crucial factor in assay validation. Ideally, PCR efficiency should reach 100%, meaning that the template doubles with each cycle. The acceptable range for efficiency is 90–110%, with an R² value greater than 0.98 on the standard curve.
For absolute quantification, intra- and inter-assay precision and accuracy must be monitored to meet predefined criteria for quality control (QC) samples with known concentrations.
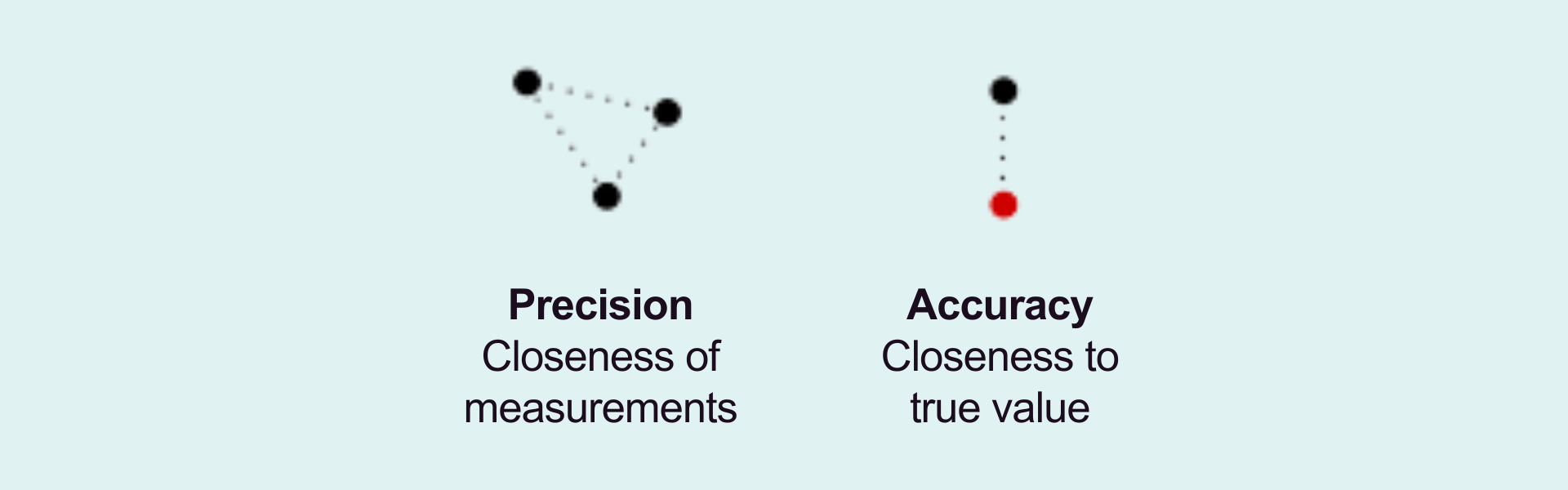
Sensitivity is a key parameter, especially when quantifying low-copy-number targets. The limit of detection (LOD) defines the lowest copy number that can be reliably detected with 95% confidence. It marks the lowest analyte concentration, producing a distinguishable signal from a no-template control (NTC). To ensure robustness, LOD validation should be performed across multiple runs. Ideally, at least three runs should be conducted by at least two analysts over at least two days. The lower limit of quantification (LLOQ) must also be verified during precision and accuracy testing.
Specificity and selectivity are evaluated during method development to confirm that the assay accurately detects the intended target without cross-reactivity or interference. Assay robustness is assessed by testing the impact of internal variations, such as changes in annealing temperature, pipetting accuracy, and reagent stability.
To ensure consistent assay performance, stability studies should account for multiple conditions, including freeze-thaw cycles, short-term and long-term storage, and temperature fluctuations.
Method validation
Precision
Accuracy
PCR efficiency
Dilutional linearity (if applicable)
Co-linearity (if applicable)
Sensitivity:
- LOB (if applicable)
- LOD
- LLOQ
Specificity
Selectivity
Robustness and Ruggedness
Stability (if applicable):
- Freeze/thaw
- Bench top
- Frozen
Extraction efficiency
Validated qPCR and dPCR assays for short RNA
Short nucleic acids, such as siRNA, miRNA, antisense oligonucleotides (ASOs), and guide RNAs (gRNA), are too short to be amplified using conventional qPCR primers. Two-Tailed PCR technology, invented by TATAA Biocenter, solves this challenge by elongating the target during the reverse transcription step, enabling a validated qPCR/dPCR approach for accurate detection and quantification of short nucleic acids in tissues and biofluids.
The Two-Tailed Technology allows even very short sequences to be analyzed with the high sensitivity and precision of qPCR and dPCR.
We have developed hundreds of custom assays using this technology, supporting applications in both research and regulated bioanalysis.
References