Genomic biomarkers

Introducing genomic biomarker analysis
A biomarker is an objectively measurable factor that can indicate disease risk, predict treatment response, or provide quantifiable evidence of treatment efficacy. Genomic biomarkers are DNA or RNA characteristics that provide insight into disease susceptibility, drug response, or biological processes that require monitoring. These can include DNA-based markers such as SNPs, haplotypes, or copy number variations, and RNA-based markers such as transcript sequences, splicing events, or gene expression levels. Beyond human samples, genomic biomarkers also extend to viruses, infectious agents, and animal samples.
The FDA classifies genomic biomarkers into categories such as exploratory, probable valid, and known valid biomarkers. Biomarker qualification involves a clinical validation, where its reliability in reflecting a biological event or clinical outcome is assessed, and the analytical validation ensures that the assay used for measurement performs reliably under defined conditions.
Key technologies for genomic biomarker analysis include qPCR, dPCR, and various next-generation sequencing (NGS) platforms. Analytical validation follows a fit-for-purpose approach per FDA guidelines, meaning validation requirements depend on the context of use (COU), ranging from highly stringent diagnostic biomarkers to more flexible exploratory research applications.
Genomic biomarkers glossary
Single nucleotide polymorphisms (SNPs) are single base pair variations commonly used as biomarkers in pharmacogenomics and disease risk assessment.
Cytogenetic rearrangements are structural changes in chromosomes, such as translocations, duplications, deletions, and inversions associated with genetic disorders and cancers.
Short sequence repeats (SSRs) are repeated DNA sequences, such as microsatellites.
Haplotypes are combinations of SNPs inherited together.
DNA modifications are epigenetic changes, such as DNA methylation, that regulate gene expression without altering the DNA sequence.
Insertions and deletions (INDELs) refer to the gain (insertion) or loss (deletion) of small DNA segments.
Copy number variations (CNVs) refer to differences in the number of copies of a particular DNA segment, often implicated in neurodevelopmental disorders, cancers, and immune diseases.
Circulating tumor DNA (ctDNA) consists of small fragments of tumor-derived DNA circulating as cell-free DNA in the bloodstream. It is used in liquid biopsies to detect tumor burden, minimal residual disease (MRD), and acquired resistance mutations in cancer therapy.
MicroRNAs (miRNAs) are small, non-coding RNAs that regulate gene expression post-transcriptionally.
RNA expression levels refer to the quantitative measurement of mRNA transcripts to assess gene activity across different conditions, diseases, and treatments.
Transgene expression refers to the expression of a therapeutic transgene in gene therapy applications, often serving as a pharmacodynamic biomarker to evaluate treatment efficacy and durability.
Gene expression profiles represent the collective measurement of gene expression across biological samples to classify disease subtypes, predict treatment response, or monitor therapeutic effects.
Regulatory guidelines for analytical validation
Accurate biomarkers are essential in drug development. A biomarker must be clinically relevant (accurately reflecting the monitored biological event) and analytically reliable (ensuring precise and trustworthy analytical measurements).
Biomarker assay validation follows a fit-for-purpose approach, where the context of use (COU) guides the analytical validation requirements. The fit-for-purpose approach means that validation can be partial or full, depending on the intended application of the biomarker assay.
The context of use (COU) should be established, including understanding the decisions based on this data. COU is the base for the validation strategy (i.e., exploratory research, pharmacodynamic monitoring, or regulatory submission), and the validation should be further refined with further information and the development of the COU.
According to ICH (The International Conference on Harmonisation) E16, the COU should be clearly outlined in the submission, specifying the biomarker’s role in drug or biotechnology product development. It is also essential to describe the assay performance, including accuracy, precision, and sample handling/storage.
The COU and the level of validation depends on the biomarker classification. Biomarkers used for regulatory decision-making require stringent validation and may follow a formal regulatory approval pathway. Biomarkers used for internal decision-making or exploratory research have more flexible validation requirements. Biomarkers used in companion diagnostics (CDx) must adhere to IVD/IVDR regulatory pathways, which are beyond the scope of this text.
In general, during the early phases of clinical studies, analytical methods are validated to ensure they are sensitive enough to detect low analyte levels, precise in their measurements, and robust.
As drug development advances, validation requirements become more rigorous to ensure that analytical data can support critical regulatory and clinical decisions, such as dose selection, efficacy assessments, and safety evaluations. For late-stage clinical trials (Phase III, pivotal trials) where the biomarker affects patient stratification or drug labeling, the FDA may require a regulated biomarker assay performed in a CLIA/CAP-certified or GCLP-compliant lab.
A Good Clinical Laboratory Practice (GCLP)-compliant laboratory is required for bioanalytical testing in clinical trials to ensure that data is traceable, reproducible, and meets regulatory expectations for drug development and approval. Pharmacodynamic and biomarker qualification studies conducted in a clinical setting should adhere to GCLP compliance to ensure data integrity and regulatory acceptance.
Complex matrices and analytes
In traditional bioanalysis, analytes, such as small molecules or biologics, are well-defined and measured in standardized matrices like plasma or blood, following strict protocols to minimize matrix effects.
In genomic biomarker assays, however, sample matrices are often diverse and highly variable, including blood, urine, cerebrospinal fluid (CSF), tissue, and saliva. This biological variability necessitates additional reference standards, normalization strategies, and stringent quality controls to ensure accurate quantification.
Regulatory guidelines generally assume stable analytes, but nucleic acids are unstable, requiring reverse transcription (RT) controls, spike-in controls, and degradation assessments to ensure data reliability.
Preanalytical factors affecting biomarker analysis: sampling and extraction
Method validation includes preanalytical steps such as sampling and extraction to ensure accuracy, robustness, and minimal variability that could affect biomarker assay performance. Different matrices contain varying levels of PCR inhibitors, and specimen-related variability can impact extraction efficiency and downstream assay performance, requiring thorough assessment and mitigation.
To meet validation criteria and optimize downstream applications, it is crucial to establish standardized collection methods (e.g., blood collection using PAXgene tubes), conduct sample integrity and stability testing, evaluate extraction efficiency and recovery rates, and implement nucleic acid quality control metrics.

qPCR and dPCR in biomarker analysis
qPCR and dPCR are highly sensitive techniques widely used to detect genomic biomarkers, particularly in gene therapy-based medicinal products and cell therapies for bioanalysis and changes in gene expression.
Both technologies amplify a target sequence, generating a fluorescent signal with each amplification cycle. However, their quantification approaches differ:
qPCR (quantitative PCR): In absolute quantification, the cycle threshold (Ct/Cq) at which the fluorescent signal reaches a defined level is compared to a standard curve with known concentrations.
dPCR (digital PCR): The sample is partitioned into thousands of individual reactions, where each partition either contains the target sequence (fluorescent signal present) or does not (no signal). The proportion of positive partitions provides an absolute digital readout of the target molecule’s concentration without needing a standard curve.
Relative qPCR: Gene expression is quantified relative to one or more endogenous housekeeping genes, selected for their stable expression across different matrices, treatment groups, and conditions to ensure reliable normalization.
Validation of qPCR and dPCR in biomarker analysis
Absolute quantification using qPCR or dPCR measures the copy number of the target in a sample. It is widely applied in biomarker quantification and bioanalysis for advanced therapies, including biodistribution, pharmacokinetics (PK), and shedding assays. In contrast, relative quantification in qPCR determines the fold change in gene expression between samples and is used for gene expression analysis, immune profiling, and pharmacodynamics.
In relative expression assays, normalization against the expression of a reference gene is critical to ensure accurate quantification. The reference gene should exhibit a stable expression profile that matches the target gene under the study conditions. Multiple reference genes enhance accuracy by compensating for biological and technical variability.
The design and validation of a PCR-based analytical assay should be driven by its context of use (COU), with the intended purpose when defining suitable acceptance criteria. Precision and accuracy must be rigorously evaluated for regulatory validation using biological samples with known template concentrations.
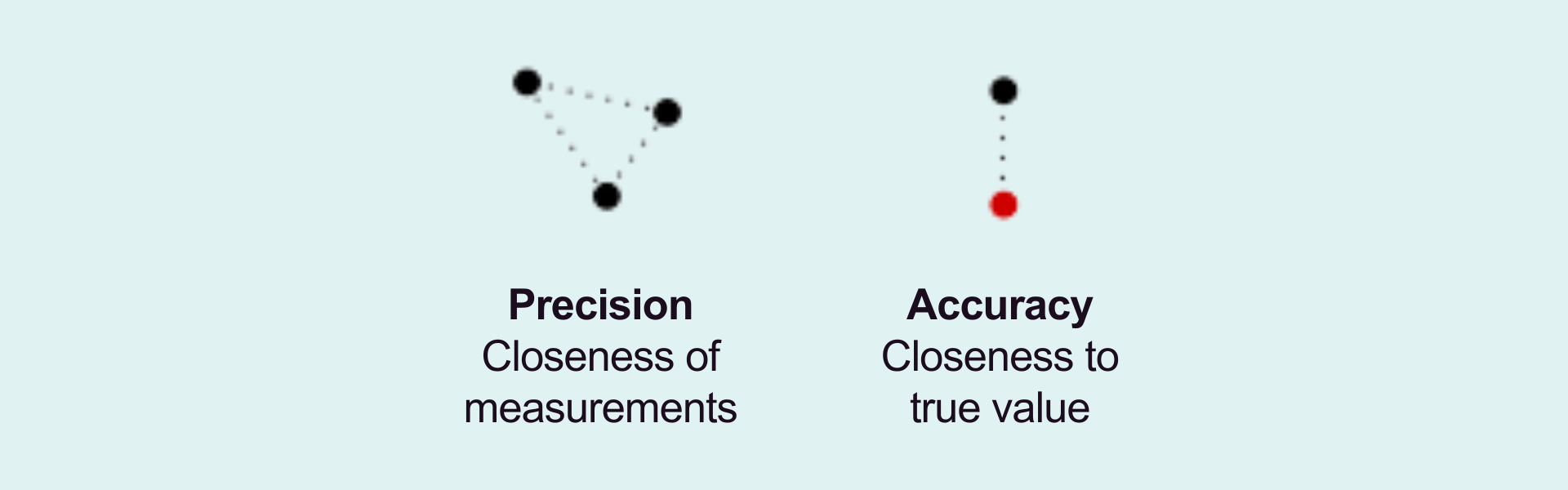
PCR efficiency is a crucial factor in assay validation. Ideally, PCR efficiency should reach 100%, meaning that the template doubles with each cycle. The acceptable range for efficiency is 90–110%, with an R² value greater than 0.98 on the standard curve. When assessing changes in gene expression relative to a housekeeping gene, the amplification efficiency of the reference gene(s) must be similar to that of the target gene. A primary consideration to ensure accuracy in relative quantification is the validation of stable reference gene expression across all experimental conditions.
For absolute quantification, intra- and inter-assay precision and accuracy must be monitored to meet predefined criteria for quality control (QC) samples with known concentrations.
Sensitivity is a key parameter, especially when quantifying low-copy-number targets. The limit of detection (LOD) defines the lowest copy number that can be reliably detected with 95% confidence. It marks the lowest analyte concentration, producing a distinguishable signal from a no-template control (NTC). To ensure robustness, LOD validation should be performed across multiple runs. Ideally, at least three runs should be conducted by at least two analysts over at least two days. The lower limit of quantification (LLOQ) must also be verified during precision and accuracy testing.
Specificity and selectivity are evaluated during method development to confirm that the assay accurately detects the intended target without cross-reactivity or interference. Assay robustness is assessed by testing the impact of internal variations, such as changes in annealing temperature, pipetting accuracy, and reagent stability.
Stability studies should account for multiple conditions, including freeze-thaw cycles, short-term and long-term storage, and temperature fluctuations, to ensure consistent assay performance.
The next-generation sequencing technology
Next-generation sequencing (NGS) uses predefined sequencing panels to enable unbiased exploration of genomic biomarkers, whether across the entire genome (whole genome sequencing), all coding regions (whole exome sequencing), or targeted gene panels. It is also widely applied to quantify the entire transcriptome (total RNA), protein-coding transcripts (messenger RNA), or specific transcript classes, such as microRNAs.
Next-generation sequencing (NGS) enables the analysis of DNA and RNA from various biological samples, provided that the extracted nucleic acids meet the required standards for quality and quantity.
During library preparation, the nucleic acids are fragmented into specific size ranges, and platform-specific adapter sequences are ligated to the ends of the fragments. These adapters contain regions complementary to the sequencing primers and often include unique barcode sequences, allowing multiple samples to be multiplexed (pooled and sequenced together) in a single run while maintaining sample identity.
Target enrichment is performed to isolate specific genes, genomic regions, or RNA targets if the
application requires it. Target enrichment is typically done using hybrid capture or amplicon-based methods, depending on the assay design.
The prepared libraries are loaded onto a sequencing platform during the sequencing step. The sequencing instrument performs massively parallel sequencing by detecting the incorporation of nucleotides as they are added to growing DNA strands. This generates millions to billions of short sequence reads per run.
The resulting raw data is processed through a bioinformatics pipeline. The bioinformatics pipeline includes base calling, identifying the nucleotide at each position in a sequencing read, and read alignment, where sequence reads are mapped to a reference genome or transcriptome.
Variant calling is then performed to detect differences compared to the reference sequence. The accuracy and sensitivity of the analysis depend on the sequencing depth (coverage). Higher coverage improves confidence in variant detection and is essential for identifying low-frequency variants, such as those found in circulating tumor DNA (ctDNA).
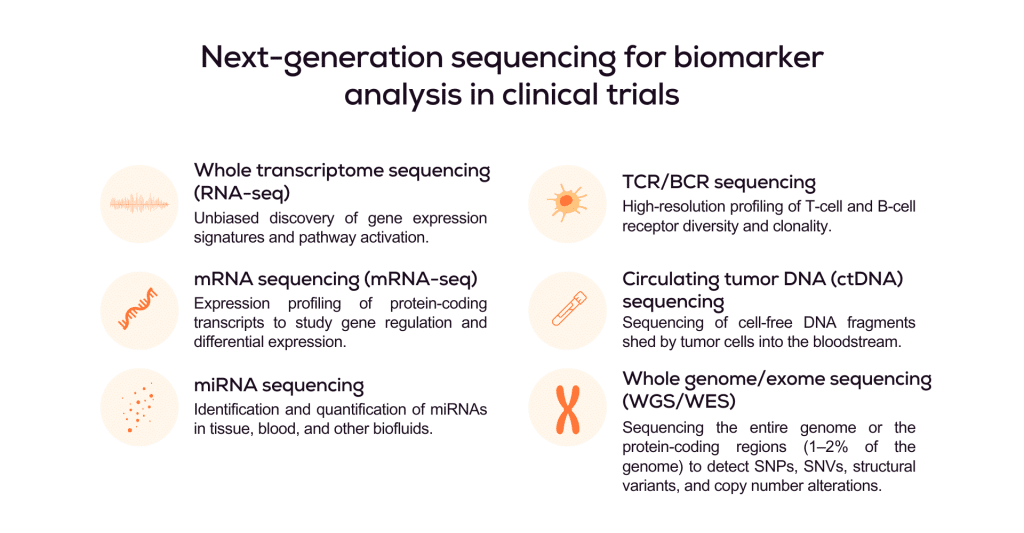
Next-generation sequencing validation for biomarker analysis
NGS is a significantly faster and higher-throughput sequencing method than targeted approaches like qPCR. However, the analytical validation process is more complex due to the increased technological and data complexity.
Like all biomarkers, the required analytical validation of a next-generation sequencing (NGS) workflow should be defined by its context of use (COU). Biomarkers intended for regulatory decision-making, such as stratification biomarkers or clinical endpoints, require more stringent validation than those used for exploratory purposes or biomarker discovery.
NGS workflows not intended for patient stratification or regulatory submissions typically follow a risk-based validation approach. This involves confirming sequencing performance metrics such as read depth, coverage uniformity, and base quality scores to ensure data reliability.
The sensitivity and dynamic range of the assay can be evaluated using dilution series of known biomarker standards. The limit of detection (LOD) and lower limit of quantification (LLOQ) are determined by assessing whether a target can be consistently detected and accurately quantified across multiple replicates and whether the observed signal is distinguishable from sequencing noise or platform error rates. These thresholds may differ depending on the application. Gene expression profiling typically requires moderate sensitivity and broader dynamic range, whereas ctDNA detection demands ultra-high sensitivity to identify low-frequency variants reliably.
The bioinformatics pipeline is also validated to ensure accurate read alignment, variant calling, and gene expression quantification, including normalization methods. Results are typically cross-validated using orthogonal techniques such as qPCR, dPCR, or Sanger sequencing.
Precision and reproducibility are assessed through intra- and inter-run testing and inter-operator reproducibility, using predefined coefficient of variation (CV) thresholds to determine acceptability.